viewer
What
The purpose of the viewer command is to provide a easy-to-use graphical-user interface to allow users to:
explore the different AncientMetagenomeDir tables via drop down menus
filter a given table based on different criteria using a standard tabular interface
download the resulting sequencing data via different methods
download prepared input samplesheets for pipelines
download citation information of selected samples
⚠️ The
viewertool was previously namedfilter, and might still be referred as such in some parts of the documentation.
For video based walkthroughs please see Tutorials.
When
You should use this tool when you wish to find particular types of ancient metagenomic data, but wish to explore the dataset manually and interactively (i.e., don’t know exactly what you’re looking for yet), and/or if you do not wish to download the AncientMetagenomeDir full tables yourself and filter them within languages such as R or Python (with pandas).
How
Loading the interface
Before using AMDirT viewer make sure you have modern web-browser (Chrome, Firefox, Edge, Safari etc.) available on your machine. This will in most cases be your own laptop or desktop - not a server.
To open the graphical user interface, open a terminal (activating software environments if necessary, see AMDirT Installation Page) and run:
AMDirT viewer
Your web-browser should automatically open a new tab and have the viewer interface open for you. If your browser does not automatically open, just copy and paste one of the URLs present in the console in your web browsers URL bar.
Local URL: http://localhost:8501
Network URL: http://172.0.0.0:8501
⚠️ The first time you run the command, you first may get some prompts in your terminal. You can just say ‘no’ to all - this will not affect the usage of the tool.
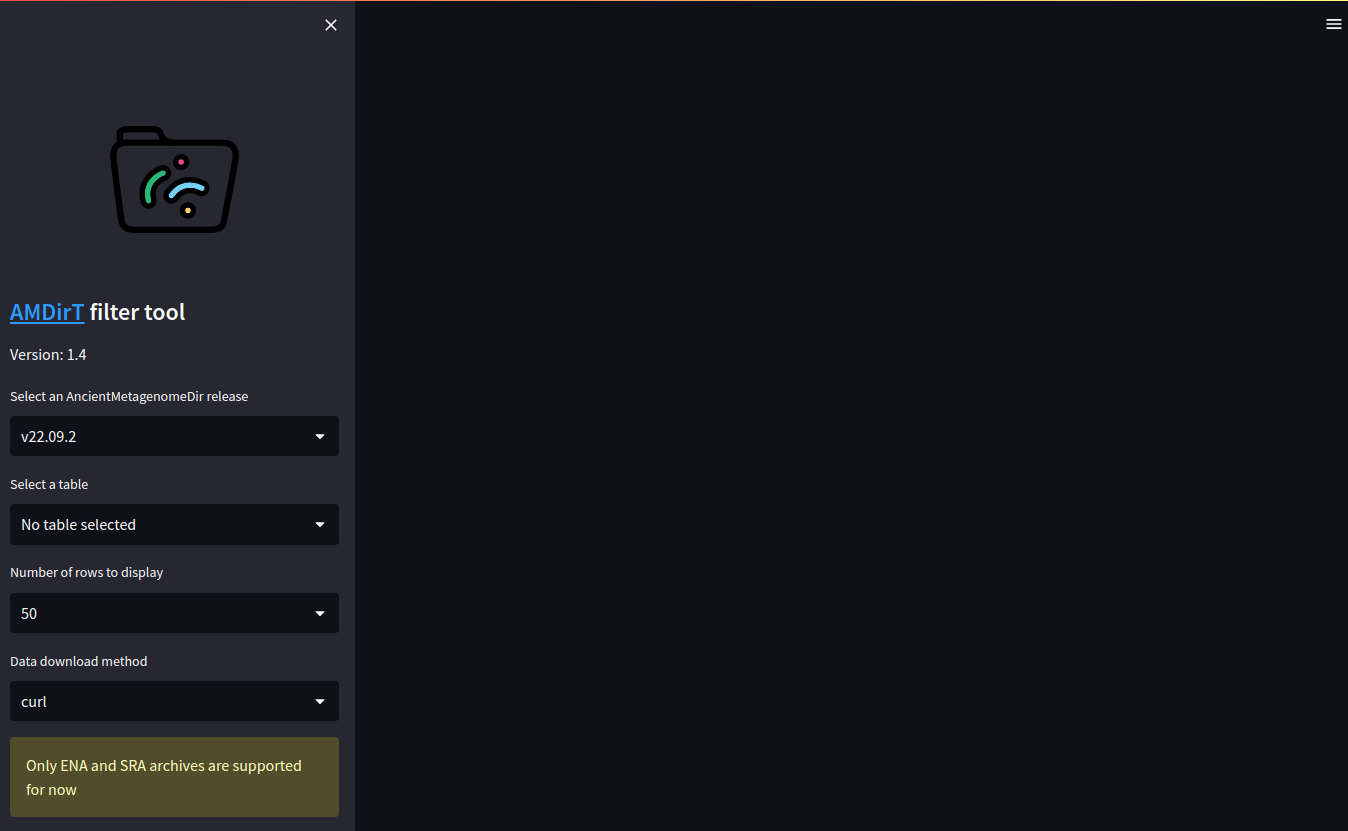
Once opened you should see a side bar and a empty pane. You can use the side bar to select various aspects of the displayed data from the AncientMetagenomeDir tables, such as the release, which table you view (e.g., environmental or host-associated metagenomics) and the number of rows to display. The data download option controls which tool the resulting download script will use to get the data.
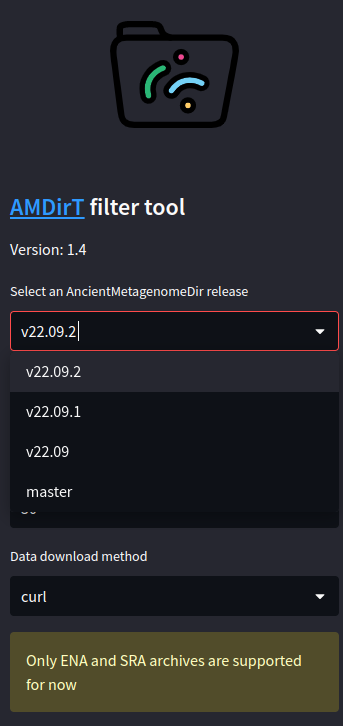
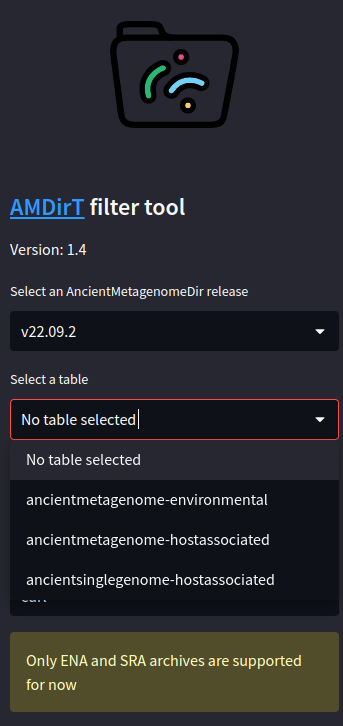
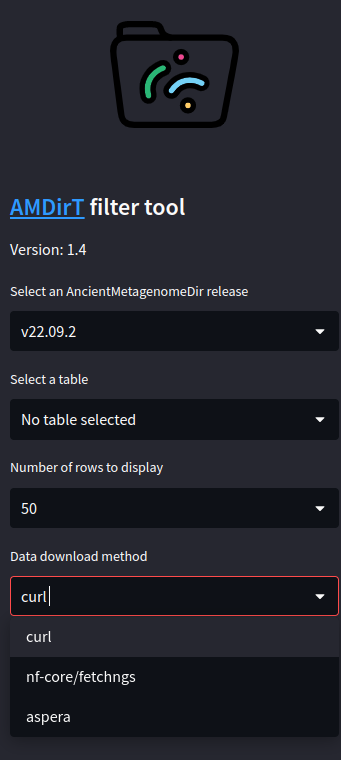
Once you have selected a version and a table, the table itself should open in the empty pane.
ℹ️ In most cases the most recent version is recommended, whereby you pick the most recent date e.g. v22.12 over v22.09 from December and September 2022 respectively
To help decide which download method to use, see the Miscellaneous page.
Exploring the Tables
To navigate the tables you can scroll up and down with your mouse. To side scroll you can hold shift and scroll. To go to the next page of samples, you can find the forward/back buttons in the bottom right of the table to navigate across pages of data entries.
To filter the tables, hover your cursor over the column of interest and you should see a ‘hamburger’ icon, press this to open the filter and table formatting options.
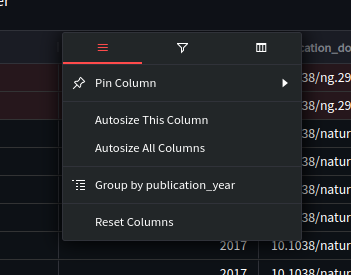


With this pane and tabs you can:
Change the size and row ordering of the column (e.g. pin this column to the left or right)
Filter the rows by various operations on this column (equal, not equal, within range, etc.)
Change which columns are displayed overall
You can also re-order the order of columns across the table by click and holding the column name, and dragging left and right.
Exporting Information
Now, select all the samples you wish to download and/or analyse.
You can select all samples currently displayed by clicking the empty box on the project_name column, or you can select individual samples by clicking the empty boxes on these rows in the project_name column.
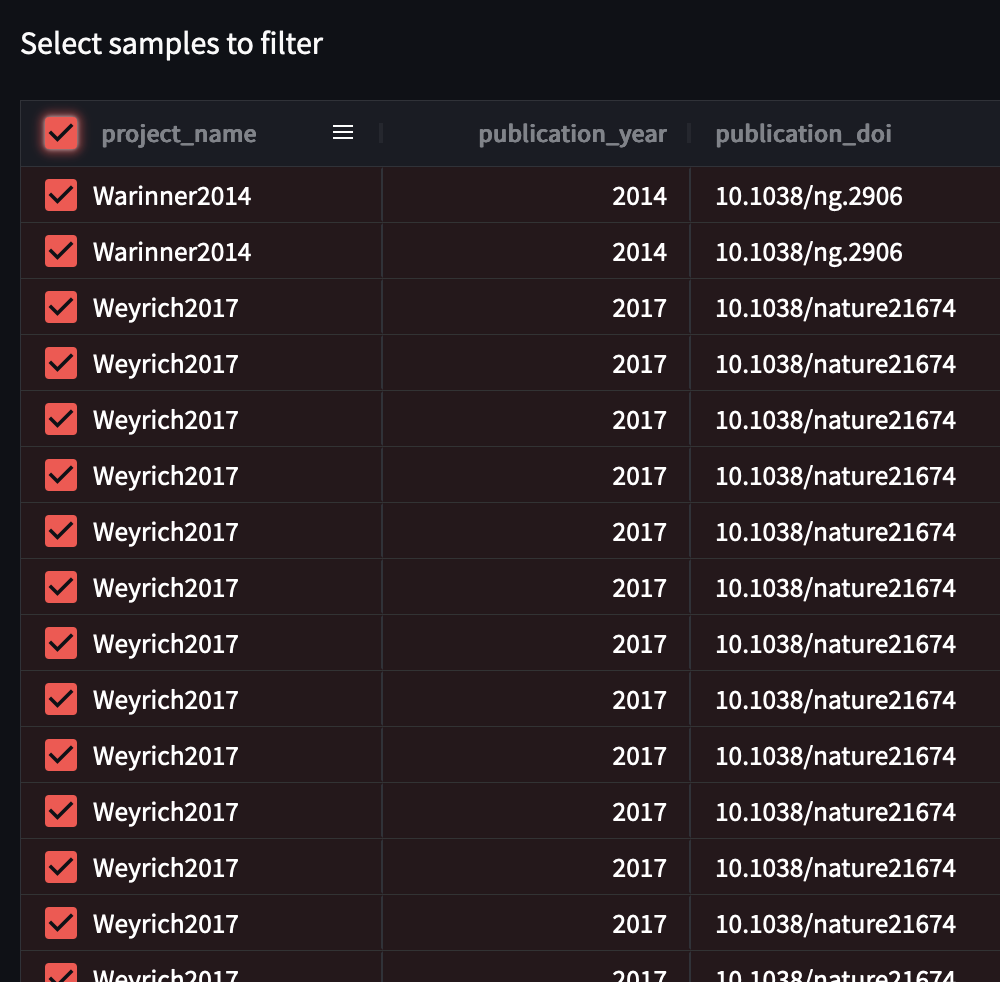

Now validate your selection! Press the ‘Validate selection’ button at the bottom of the table.
⚠️ If you wish to download the data, make sure you have already selected your ‘Data download method’ in the sidebar before pressing ‘Validate selection’!
Once the selection is validated, the library filtering table will appear below
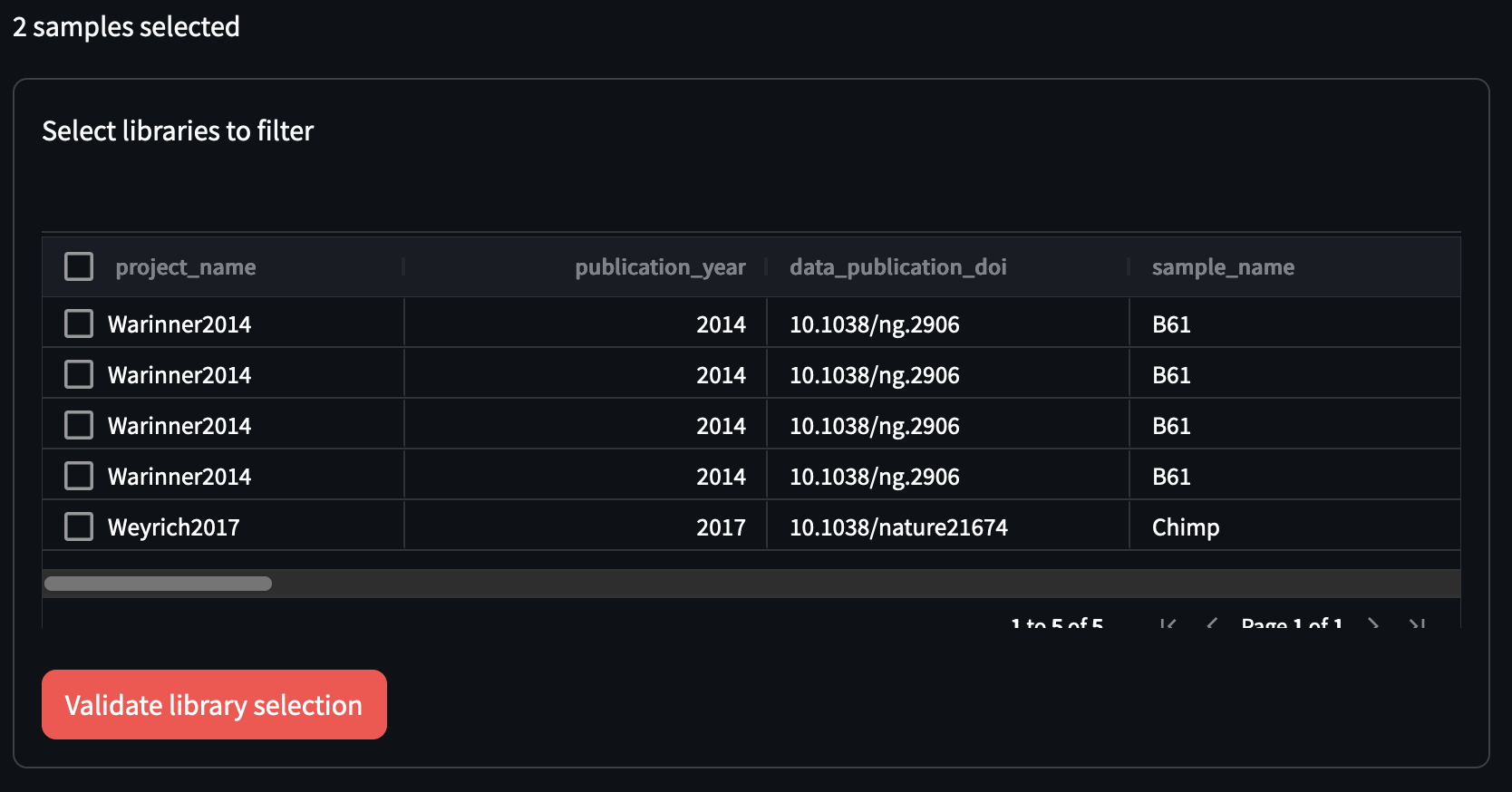
You can proceed to select your libraries of choice (or all of them) in the same way as the samples table. Once happy, you can then click on ‘Validate library selection’
Your different download options will finally appear:
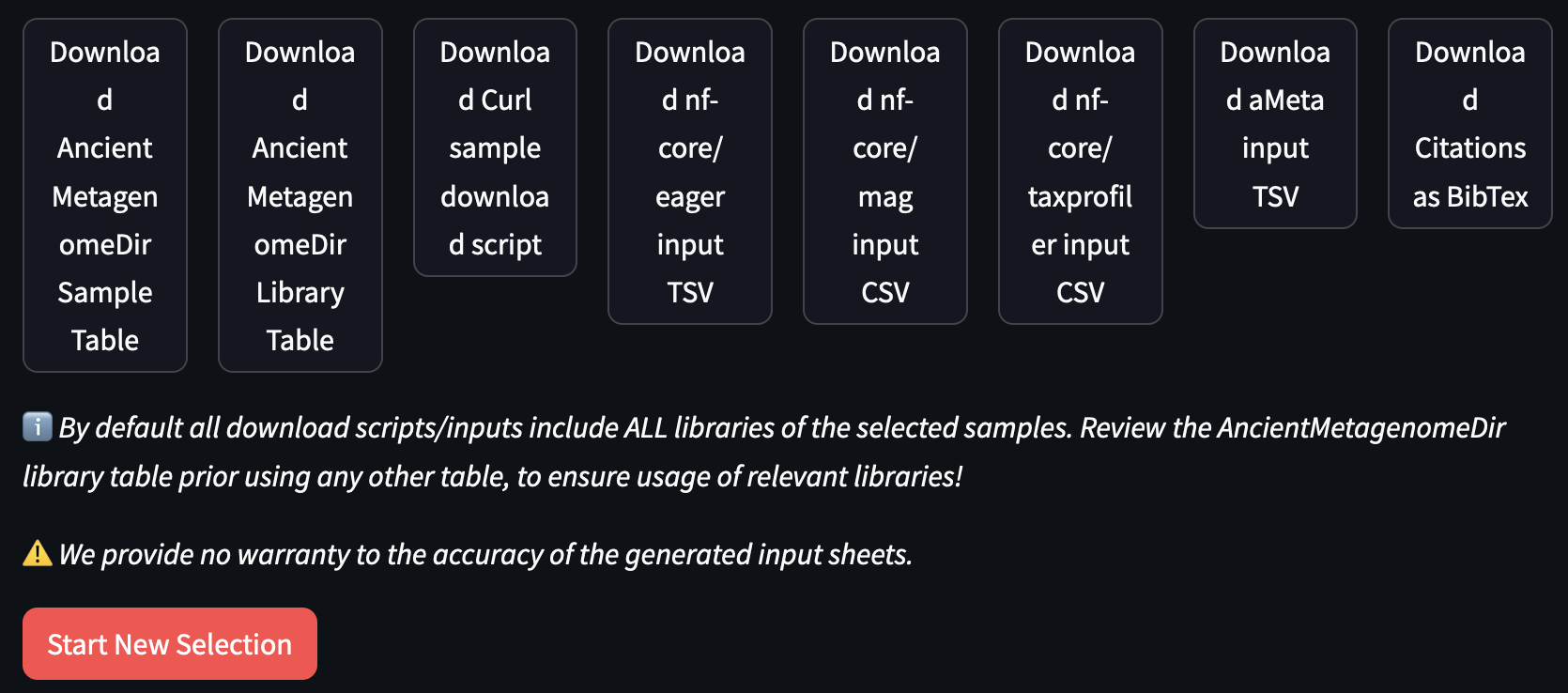
In this case we suggest you press:
Download Curl sample download script: contains a bash script with download commands for all the sequencing libraries of the selected samples
Download nf-core/eager input/tsv: a pipeline input samplesheet: a pre-configured input sheet for a pipeline, based on the downloaded files
Download citations as BibTex: citation information in BibTex format for selected samples
See Output for descriptions of all output possible files.
To use the download script, you can simply run:
bash ancientMetagenomeDir_curl_download_script.sh
⚠️ We highly recommend downloading and reviewing
AncientMetagenomeDir_filtered_libraries.tsvbefore running the curl download script to ensure you have in the download scripts and/or pipeline input sheets only the actual library types you wish to use (e.g. you may only want paired-end data, or non-UDG treated data).
The sequencing files of the selected samples will then have been downloaded to the directory you run the script from.
⚠️ You must already have the download tool you have selected installed and configured on your machine to use the bash script!
⚠️ Make sure you only run this script on the machine you will run your analyses from, i.e. in most cases on a server or HPC!
To use the pipeline input samplesheet, you should always double check the sheet is correctly configured. Once you have validated it, you can supply it to your selected pipeline as follows (using nf-core/eager as an example):
nextflow run nf-core/eager <...> --input ancientMetagenomeDir_eager_input.csv
The citations bibtex file contains all the citation information of your selected samples in a widely used format called BibTex. You can import this file into most referencing or bibliography managing tools (Zotero, Paperpile etc.).
⚠️ Occasionally the cross-ref databases do not have citation information for certain publications. You will receive a warning if so, with the relevant DOIs for you to manually get this information.
Finishing
If you have finished your selection and file downloading, you can close the interface simply by closing the tab, and then pressing Ctrl + c on your terminal.
If you wish to generate a new selection in the same session, you must press the ‘Start New Selection’ button at the bottom of the interface, select the new samples, and press ‘Validate selection’ again. If you do not press ‘Start New Selection’, you will export the same set of files and samples from your first selection.
Output
⚠️ We highly recommend generating and reviewing
AncientMetagenomeDir_filtered_libraries.tsvbefore downloading or running any pipelines to ensure you have in the download scripts and/or pipeline input sheets only the actual library types you wish to use (e.g. you may only want paired-end data, or non-UDG treated data).
⚠️ To use a pipeline input samplesheet, you should always double check the sheet is correctly configured. We cannot guarantee accuracy between metadata and sequencing files.
All possible output is as follows:
<outdir>: where all the pipeline samplesheets are placed (by default.)AncientMetagenomeDir_bibliography.bib:A BibTex format citation information file with all references (where available) present in the filtered sample table.
AncientMetagenomeDir_filtered_libraries.tsv:The associated AncientMetagenomeDir curated metadata for all libraries of the samples in the input table.
AncientMetagenomeDir_curl_download_script.sh:A bash script containing curl commands for all libraries in the input samples list.
AncientMetagenomeDir_aspera_download_script.sh:A bash script containing Aspera commands for all libraries in the input samples list. See How Tos for Aspera configuration information.
AncientMetagenomeDir_nf_core_fetchngs_input_table.tsv:An input sheet containing ERS/SRS accession numbers in a format compatible with the nf-core/fetchngs input samplesheet.
AncientMetagenomeDir_nf_core_eager_input_table.tsv:An input sheet with metadata in a format compatible with the nf-core/eager input samplesheet.
Contained paths are relative to the directory output when using the
curlandasperadownload scripts (i.e., input sheet assumes files are in the same directory as the input sheet itself).
AncientMetagenomeDir_nf_core_taxprofiler_input_table.csv:An input sheet with metadata in a format compatible with the nf-core/taxprofiler input samplesheet.
Contained paths are relative to the directory output when using the
curlandasperadownload scripts (i.e., input sheet assumes files are in the same directory as the input sheet itself).
AncientMetagenomeDir_aMeta_input_table.tsv:An input sheet with metadata in a format compatible with the aMeta input samplesheet.
Contained paths are relative to the directory output when using the
curlandasperadownload scripts (i.e., input sheet assumes files are in the same directory as the input sheet itself).⚠️ As of version/commit 1a03f3d (and earlier) aMeta does not support paired-end data! For paired-end data you must merge pairs yourself manually before using the sample sheet.
AncientMetagenomeDir_mag_input.zip:Input sheets in a format compatible with the nf-core/mag input samplesheet.
nf-core/mag does not support paired- and single-end data in the same run, therefore paired- and/or input sheets are downloaded as a Zip file that must be extracted before being supplied to the pipeline.
Contained paths are relative to the directory output when using the
curlandasperadownload scripts (i.e., input sheet assumes files are in the same directory as the input sheet itself).